今回は興味を持ってもらえる人が限定される記事だと思うけど、以前作ったコードを腐らせてしまうのも勿体無いので敢えて晒してみる。
生体内のタンパク質構造をX線回折やNMRで解析したデータの多くはPDB (Protein Data Bank)データとして登録される。以下に示すように原子タグ、原子の通し番号、原子名、アミノ酸名、アミノ酸の通し番号、原子の座標などがテキストで書かれている。そして、機能の解明などのために分子動力学(MD; molecular dynamics)計算プログラムなどでそれらのPDBデータは利用される。MD計算プログラムは世に沢山あるが、自分がよく使っていたのはAmberというソフトウェアだ。
PDBデータの一部
ATOM 1439 N ILE 212 52.408 -3.243 59.013 1.00 40.03 N ATOM 1440 CA ILE 212 52.511 -3.151 57.556 1.00 33.72 C ATOM 1441 C ILE 212 51.464 -2.243 56.911 1.00 34.65 C ATOM 1442 O ILE 212 51.733 -1.619 55.886 1.00 38.03 O ATOM 1443 CB ILE 212 52.482 -4.572 56.921 1.00 38.88 C ATOM 1444 CG1 ILE 212 53.615 -4.703 55.919 1.00 40.75 C ATOM 1445 CG2 ILE 212 51.129 -4.901 56.281 1.00 39.55 C ATOM 1446 CD1 ILE 212 54.931 -4.446 56.550 1.00 46.31 C ATOM 1447 N HIS 213 50.308 -2.142 57.570 1.00 36.88 N ATOM 1448 CA HIS 213 49.160 -1.334 57.169 1.00 37.60 C ATOM 1449 C HIS 213 49.523 0.121 56.879 1.00 39.63 C ATOM 1450 O HIS 213 48.887 0.773 56.028 1.00 39.63 O ATOM 1451 CB HIS 213 48.115 -1.389 58.289 1.00 44.39 C ATOM 1452 CG HIS 213 46.795 -0.746 57.947 1.00 55.55 C ATOM 1453 ND1 HIS 213 46.364 -0.536 56.651 1.00 54.19 N ATOM 1454 CD2 HIS 213 45.800 -0.306 58.748 1.00 53.97 C ATOM 1455 CE1 HIS 213 45.166 0.002 56.670 1.00 51.63 C ATOM 1456 NE2 HIS 213 44.787 0.157 57.930 1.00 44.85 N ATOM 1457 N CYS 214 50.530 0.634 57.584 1.00 37.10 N ATOM 1458 CA CYS 214 50.986 2.011 57.385 1.00 37.27 C ATOM 1459 C CYS 214 51.504 2.255 55.949 1.00 38.90 C ATOM 1460 O CYS 214 51.699 3.400 55.518 1.00 39.15 O ATOM 1461 CB CYS 214 52.037 2.389 58.443 1.00 36.90 C ATOM 1462 SG CYS 214 53.736 1.793 58.215 1.00 34.05 S
タンパク質はアミノ酸の集合体である。アミノ酸の分子構造はよく知られているが、一部のアミノ酸については水素が結合する位置が異なることがある。例えばヒスチジン(HIS)というアミノ酸では、以下の3種が存在することが知られている(図中の3文字記号はAmberで使われる名称)。
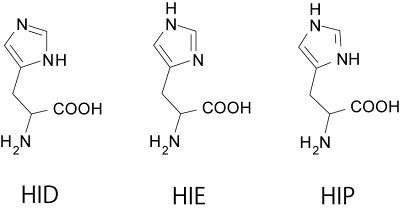
ここでタンパク質に対するMD計算について考えてみる。MD計算ではアミノ酸ごとに計算のためのデータベースを持っており、それを利用して計算される。なので、同じヒスチジンでも水素の付加の位置によって異なるデータが使われる。しかし、PDBデータの多くは水素が付加されておらず(X線結晶構造解析では水素の位置は特定できない)、計算前にアミノ酸の配置や想定されるpHなどから水素を付加させる必要がある。そして、ヒスチジン毎に異なる位置に水素が付加されるわけだ。
異なる位置に水素が付加されたヒスチジンに対して、正しい計算データをMD計算プログラムに伝えるためにそのアミノ酸の名前を変更する必要があるのだが、これが結構な手間となる。例えば、1000個以上のタンパク質構造からヒスチジン分子をビューアで確認してその水素の位置からアミノ酸の名前を付け替えるという作業なんて誰もやりたくないだろう。
そこで、水素の位置の異なるアミノ酸を自動的に判定してアミノ酸の名前を付け直すプログラムを作った。このコードでは部分グラフ同型判定アルゴリズムを利用している。使用したアルゴリズムをここで説明するのは面倒なので、興味のある方はV. Lipets, N. Vanetik, and E. Gudes. Subsea: an efficient heuristic algorithm for subgraph isomorphism. Data Min. Knowl. Disc. 19. 320-350 (2009)あたりを参考にしてほしい。
使い方は次の通り。以下に示す例では、input.pdbが対象とするPDBデータで、名前がHISとなっているアミノ酸について判定する。判定するためのリファレンス構造は3種類で、HID.pdb、HIE.pdb, HIP.pdbの3つのPDB構造に対して行う。構造と一致したら、それぞれアミノ酸名をHID、HIE、HIPに置き換える。最終的に修正した構造をoutput.pdbとして出力する。
% matching_residue input.pdb output.pdb HIS HID HID.pdb HIE HIE.pdb HIP HIP.pdb
標準出力には以下のような表示が出る。この表示の場合、入力PDBファイルには6個のHISがあり、それぞれ17, 66, 132, 146, 167, 261番目のアミノ酸で、HIEとHIPに変換されたことを示している。ここでは判定するための対象としてヒスチジンを例に取っているが、アスパラギン酸やリシンでも構わないし、アミノ酸でなくてもいい。
Number of HIS: 6 HIS 17 -> HIE HIS 66 -> HIE HIS 132 -> HIE HIS 146 -> HIP HIS 167 -> HIE HIS 261 -> HIP
今回に処理については、必ずしも部分グラフとして判定する必要はないのだが、今後の拡張のため、例えばアミノ酸の一部から正しい構造を判別する場合などについても考慮して部分グラフとして扱えるようにしている。
最後に今回のプログラムのソースコードを示す。
matching_residue.cpp
// matching_residue.cpp by nox, 2011.04.18 #include <iostream> #include <fstream> #include <sstream> #include <string> #include <vector> #include <map> #include <algorithm> #include <utility> #include <iomanip> #include <limits> #include <cmath> using namespace std; struct Node { vector<Node*> nb; // Node list of neighbor atoms ([Node]) int idx; // index of the atom in the residue (int) int d; // index of traverse history (int) vector<int> t; // neighbors of traverse history ([int]) string atom; // atom type (string) int res_id; // residue id of the PDB (int) vector<double> crd; // coordinate of the atom ([float,float,float]) Node(int idx_, string atom_, int res_id_, vector<double>& crd_) : idx(idx_), d(-1), atom(atom_), res_id(res_id_), crd(crd_) { } }; struct PdbData { string atom; int res_id; vector<double> crd; PdbData(string line) { stringstream ss; double d; atom = line.substr(76, 2); atom.erase(0, atom.find_first_not_of(' ')); atom.erase(atom.find_last_not_of(' ') + 1); ss << line.substr(20, 6); ss >> res_id; ss.str(""); ss.clear(); ss << line.substr(30, 8); ss >> d; crd.push_back(d); ss.str(""); ss.clear(); ss << line.substr(38, 8); ss >> d; crd.push_back(d); ss.str(""); ss.clear(); ss << line.substr(46, 8); ss >> d; crd.push_back(d); ss.str(""); ss.clear(); } }; // comparison function for sorting of traverse history class LessNeighborD { public: bool operator()(const Node* x, const Node* y) { int x_min_d, y_min_d; x_min_d = y_min_d = numeric_limits<int>::max(); for (vector<Node*>::const_iterator p = x->nb.begin(); p != x->nb.end(); ++p) { if ((*p)->d == -1) continue; x_min_d = min(x_min_d, (*p)->d); } for (vector<Node*>::const_iterator p = y->nb.begin(); p != y->nb.end(); ++p) { if ((*p)->d == -1) continue; y_min_d = min(y_min_d, (*p)->d); } return x_min_d < y_min_d; } }; class GreaterNbSize { public: bool operator()(const Node* x, const Node* y) { return x->nb.size() > y->nb.size(); } }; class LessD { public: bool operator()(const Node* x, const Node* y) { return x->d < y->d; } }; class Matching { private: // visit time of traverse history int vtime; // mapping between target and the replacing residue ((Node,Node)) vector<pair<Node*, Node*> > mapping; map<pair<string, string>, double> bond_dist; public: Matching(); double get_bond_dist(const Node* v1, const Node* v2); double distance(const Node* v1, const Node* v2); void connect(vector<Node*>& nodes); void traverse_history(Node* node); void clear_traverse_history(vector<Node*>& nodes); vector<pair<string, vector<string> > > parse_pdb(string inpdb); vector<vector<Node*> > create_nodes_list(vector<pair<string, vector<string> > >& all_lines, vector<string>& rep_res); bool match(Node* g, vector<Node*>& h, int depth); }; Matching::Matching() : vtime(0) { // table of max bond distances bond_dist[make_pair("C", "C")] = 2.0; bond_dist[make_pair("C", "N")] = 1.8; bond_dist[make_pair("C", "O")] = 1.8; bond_dist[make_pair("C", "H")] = 1.2; bond_dist[make_pair("N", "N")] = 1.8; bond_dist[make_pair("N", "O")] = 1.8; bond_dist[make_pair("H", "N")] = 1.2; bond_dist[make_pair("O", "O")] = 1.8; bond_dist[make_pair("H", "O")] = 1.2; } double Matching::get_bond_dist(const Node* v1, const Node* v2) { pair<string, string> atom_pair(make_pair(min(v1->atom, v2->atom), max(v1->atom, v2->atom))); if (bond_dist.find(atom_pair) != bond_dist.end()) return bond_dist[atom_pair]; return 0.0; } double Matching::distance(const Node* v1, const Node* v2) { double d = 0.0; for (int i = 0; i < 3; i++) { double dv = v1->crd[i] - v2->crd[i]; d += dv * dv; } return sqrt(d); } // create connect list for neighbor atoms void Matching::connect(vector<Node*>& nodes) { for (vector<Node*>::iterator p = nodes.begin(); p != nodes.end(); ++p) { for (vector<Node*>::iterator q = nodes.begin(); q != nodes.end(); ++q) { if (*p == *q || (*p)->idx > (*q)->idx) continue; if (distance(*p, *q) < get_bond_dist(*p, *q)) { (*p)->nb.push_back(*q); (*q)->nb.push_back(*p); } } } } // traverse history, similar to canonical labeling void Matching::traverse_history(Node* node) { node->d = vtime; vtime++; vector<Node*> nb; for (vector<Node*>::iterator p = node->nb.begin(); p != node->nb.end(); ++p) { if ((*p)->d == -1) nb.push_back(*p); else node->t.push_back((*p)->d); } sort(node->t.begin(), node->t.end()); sort(nb.begin(), nb.end(), LessNeighborD()); for (vector<Node*>::iterator p = nb.begin(); p != nb.end(); ++p) if ((*p)->d == -1) traverse_history(*p); } void Matching::clear_traverse_history(vector<Node*>& nodes) { for (vector<Node*>::iterator p = nodes.begin(); p != nodes.end(); ++p) { (*p)->d = -1; (*p)->t.clear(); } mapping.clear(); } vector<pair<string, vector<string> > > Matching::parse_pdb(string inpdb) { int res_id, prev_res_id = -1; vector<string> lines; vector<pair<string, vector<string> > > all_lines; string res, line; stringstream ss; fstream fs(inpdb.c_str(), ios_base::in); while (getline(fs, line)) { ss.str(""); ss.clear(); if (line.length() > 20 && line.substr(0, 4) == "ATOM") { ss << line.substr(20, 6); ss >> res_id; } else res_id = -1; if (prev_res_id == res_id) lines.push_back(line); else { if (!lines.empty()) all_lines.push_back(make_pair(res, lines)); lines.clear(); lines.push_back(line); } if (line.length() > 20 && line.substr(0, 4) == "ATOM") { res = line.substr(17, 3); res.erase(0, res.find_first_not_of(' ')); res.erase(res.find_last_not_of(' ') + 1); } else res = ""; prev_res_id = res_id; } all_lines.push_back(make_pair(res, lines)); return all_lines; } vector<vector<Node*> > Matching::create_nodes_list(vector<pair<string, vector<string> > >& all_lines, vector<string>& rep_res) { vector<vector<Node*> > nodes_list; vector<vector<PdbData> > pdb_data; for (vector<pair<string, vector<string> > >::iterator p = all_lines.begin(); p != all_lines.end(); ++p) { string res(p->first); vector<PdbData> pdbs; for (vector<string>::iterator q = p->second.begin(); q != p->second.end(); ++q) { // for l in lines if (q->length() > 78 && q->substr(0, 4) == "ATOM" && find(rep_res.begin(), rep_res.end(), res) != rep_res.end()) pdbs.push_back(PdbData(*q)); } pdb_data.push_back(pdbs); } for (vector<vector<PdbData> >::iterator p = pdb_data.begin(); p != pdb_data.end(); ++p) { if (p->empty()) continue; vector<Node*> nodes; int idx = 0; for (vector<PdbData>::iterator q = p->begin(); q != p->end(); ++q) nodes.push_back(new Node(idx++, q->atom, q->res_id, q->crd)); connect(nodes); vtime = 0; traverse_history(nodes[0]); sort(nodes.begin(), nodes.end(), LessD()); nodes_list.push_back(nodes); } return nodes_list; } bool Matching::match(Node* g, vector<Node*>& h, int depth) { for (vector<Node*>::iterator p = g->nb.begin(); p != g->nb.end(); ++p) if ((*p)->d != -1) g->t.push_back((*p)->d); if (h[depth]->t.size() != g->t.size() || h[depth]->atom != g->atom) { g->t.clear(); return false; } g->d = depth; sort(g->t.begin(), g->t.end()); if (g->t == h[depth]->t) { mapping.push_back(make_pair(h[depth], g)); if (mapping.size() == h.size()) return true; vector<Node*> nb(mapping[h[depth+1]->t[0]].second->nb); sort(nb.begin(), nb.end()); vector<Node*>::iterator end = nb.end(); for (int i = 1; i < h[depth+1]->t.size(); i++) { vector<Node*> nb2(mapping[h[depth+1]->t[i]].second->nb); sort(nb2.begin(), nb2.end()); end = set_intersection(nb.begin(), end, nb2.begin(), nb2.end(), nb.begin()); } nb.erase(end, nb.end()); sort(nb.begin(), nb.end(), GreaterNbSize()); for (vector<Node*>::iterator p = nb.begin(); p != nb.end(); ++p) if ((*p)->d == -1 && match(*p, h, depth + 1)) return true; mapping.pop_back(); } g->d = -1; g->t.clear(); return false; } int main(int argc, char* argv[]) { string inpdb, outpdb, target; vector<pair<string, string> > res_pdb_list; if (argc < 6 || (argc & 1)) { cerr << "Usage: " << argv[0] << " input_pdb output_pdb target_res replace_res1 replace_res1_pdb [replace_res2 replace_res2_pdb...]" << endl; cerr << " ex.) " << argv[0] << " input.pdb output.pdb HIS HID HID.pdb HIE HIE.pdb HIP HIP.pdb" << endl; exit(1); } inpdb = argv[1]; outpdb = argv[2]; target = argv[3]; for (int i = 4; i < argc; i += 2) res_pdb_list.push_back(make_pair(argv[i], argv[i+1])); Matching m; // calculate nodes of replacing residues map<string, vector<Node*> > rep_res; for (vector<pair<string, string> >::iterator p = res_pdb_list.begin(); p != res_pdb_list.end(); ++p) { vector<pair<string, vector<string> > > all_lines(m.parse_pdb(p->second)); vector<string> res; res.push_back(p->first); vector<vector<Node*> > nodes_list(m.create_nodes_list(all_lines, res)); rep_res[p->first] = nodes_list[0]; } // calculate nodes of target residues in the PDB vector<pair<string, vector<string> > > all_lines(m.parse_pdb(inpdb)); vector<string> res; res.push_back(target); vector<vector<Node*> > nodes_list(m.create_nodes_list(all_lines, res)); cout << "Number of " << target << ": " << nodes_list.size() << endl; // matching residues vector<string> result; bool is_found = false; for (vector<vector<Node*> >::iterator p = nodes_list.begin(); p != nodes_list.end(); ++p) { is_found = false; for (map<string, vector<Node*> >::iterator q = rep_res.begin(); q != rep_res.end(); ++q) { if (p->size() != q->second.size()) continue; m.clear_traverse_history(*p); for (vector<Node*>::iterator r = p->begin(); r != p->end(); ++r) { if (m.match(*r, q->second, 0)) { cout << target << " " << setw(4) << (*r)->res_id << " -> " << q->first << endl; result.push_back(q->first); is_found = true; break; } } if (is_found) break; } if (!is_found) { cout << target << " " << setw(4) << (*p)[0]->res_id << " -> Not found." << endl; result.push_back(target); } } // write PDB fstream fs(outpdb.c_str(), ios_base::out); int idx = 0; for (vector<pair<string, vector<string> > >::iterator p = all_lines.begin(); p != all_lines.end(); ++p) { if (p->first == target) { for (vector<string>::iterator q = p->second.begin(); q != p->second.end(); ++q) { size_t pos = q->find(target); if (pos != string::npos) q->replace(pos, target.size(), result[idx]); fs << *q << endl; } idx++; } else { for (vector<string>::iterator q = p->second.begin(); q != p->second.end(); ++q) fs << *q << endl; } } return 0; }
生体内のタンパク質構造をX線回折やNMRで解析したデータの多くはPDB (Protein Data Bank)データとして登録される。以下に示すように原子タグ、原子の通し番号、原子名、アミノ酸名、アミノ酸の通し番号、原子の座標などがテキストで書かれている。そして、機能の解明などのために分子動力学(MD; molecular dynamics)計算プログラムなどでそれらのPDBデータは利用される。MD計算プログラムは世に沢山あるが、自分がよく使っていたのはAmberというソフトウェアだ。
PDBデータの一部
ATOM 1439 N ILE 212 52.408 -3.243 59.013 1.00 40.03 N ATOM 1440 CA ILE 212 52.511 -3.151 57.556 1.00 33.72 C ATOM 1441 C ILE 212 51.464 -2.243 56.911 1.00 34.65 C ATOM 1442 O ILE 212 51.733 -1.619 55.886 1.00 38.03 O ATOM 1443 CB ILE 212 52.482 -4.572 56.921 1.00 38.88 C ATOM 1444 CG1 ILE 212 53.615 -4.703 55.919 1.00 40.75 C ATOM 1445 CG2 ILE 212 51.129 -4.901 56.281 1.00 39.55 C ATOM 1446 CD1 ILE 212 54.931 -4.446 56.550 1.00 46.31 C ATOM 1447 N HIS 213 50.308 -2.142 57.570 1.00 36.88 N ATOM 1448 CA HIS 213 49.160 -1.334 57.169 1.00 37.60 C ATOM 1449 C HIS 213 49.523 0.121 56.879 1.00 39.63 C ATOM 1450 O HIS 213 48.887 0.773 56.028 1.00 39.63 O ATOM 1451 CB HIS 213 48.115 -1.389 58.289 1.00 44.39 C ATOM 1452 CG HIS 213 46.795 -0.746 57.947 1.00 55.55 C ATOM 1453 ND1 HIS 213 46.364 -0.536 56.651 1.00 54.19 N ATOM 1454 CD2 HIS 213 45.800 -0.306 58.748 1.00 53.97 C ATOM 1455 CE1 HIS 213 45.166 0.002 56.670 1.00 51.63 C ATOM 1456 NE2 HIS 213 44.787 0.157 57.930 1.00 44.85 N ATOM 1457 N CYS 214 50.530 0.634 57.584 1.00 37.10 N ATOM 1458 CA CYS 214 50.986 2.011 57.385 1.00 37.27 C ATOM 1459 C CYS 214 51.504 2.255 55.949 1.00 38.90 C ATOM 1460 O CYS 214 51.699 3.400 55.518 1.00 39.15 O ATOM 1461 CB CYS 214 52.037 2.389 58.443 1.00 36.90 C ATOM 1462 SG CYS 214 53.736 1.793 58.215 1.00 34.05 S
タンパク質はアミノ酸の集合体である。アミノ酸の分子構造はよく知られているが、一部のアミノ酸については水素が結合する位置が異なることがある。例えばヒスチジン(HIS)というアミノ酸では、以下の3種が存在することが知られている(図中の3文字記号はAmberで使われる名称)。

ここでタンパク質に対するMD計算について考えてみる。MD計算ではアミノ酸ごとに計算のためのデータベースを持っており、それを利用して計算される。なので、同じヒスチジンでも水素の付加の位置によって異なるデータが使われる。しかし、PDBデータの多くは水素が付加されておらず(X線結晶構造解析では水素の位置は特定できない)、計算前にアミノ酸の配置や想定されるpHなどから水素を付加させる必要がある。そして、ヒスチジン毎に異なる位置に水素が付加されるわけだ。
異なる位置に水素が付加されたヒスチジンに対して、正しい計算データをMD計算プログラムに伝えるためにそのアミノ酸の名前を変更する必要があるのだが、これが結構な手間となる。例えば、1000個以上のタンパク質構造からヒスチジン分子をビューアで確認してその水素の位置からアミノ酸の名前を付け替えるという作業なんて誰もやりたくないだろう。
そこで、水素の位置の異なるアミノ酸を自動的に判定してアミノ酸の名前を付け直すプログラムを作った。このコードでは部分グラフ同型判定アルゴリズムを利用している。使用したアルゴリズムをここで説明するのは面倒なので、興味のある方はV. Lipets, N. Vanetik, and E. Gudes. Subsea: an efficient heuristic algorithm for subgraph isomorphism. Data Min. Knowl. Disc. 19. 320-350 (2009)あたりを参考にしてほしい。
使い方は次の通り。以下に示す例では、input.pdbが対象とするPDBデータで、名前がHISとなっているアミノ酸について判定する。判定するためのリファレンス構造は3種類で、HID.pdb、HIE.pdb, HIP.pdbの3つのPDB構造に対して行う。構造と一致したら、それぞれアミノ酸名をHID、HIE、HIPに置き換える。最終的に修正した構造をoutput.pdbとして出力する。
% matching_residue input.pdb output.pdb HIS HID HID.pdb HIE HIE.pdb HIP HIP.pdb
標準出力には以下のような表示が出る。この表示の場合、入力PDBファイルには6個のHISがあり、それぞれ17, 66, 132, 146, 167, 261番目のアミノ酸で、HIEとHIPに変換されたことを示している。ここでは判定するための対象としてヒスチジンを例に取っているが、アスパラギン酸やリシンでも構わないし、アミノ酸でなくてもいい。
Number of HIS: 6 HIS 17 -> HIE HIS 66 -> HIE HIS 132 -> HIE HIS 146 -> HIP HIS 167 -> HIE HIS 261 -> HIP
今回に処理については、必ずしも部分グラフとして判定する必要はないのだが、今後の拡張のため、例えばアミノ酸の一部から正しい構造を判別する場合などについても考慮して部分グラフとして扱えるようにしている。
最後に今回のプログラムのソースコードを示す。
matching_residue.cpp
// matching_residue.cpp by nox, 2011.04.18 #include <iostream> #include <fstream> #include <sstream> #include <string> #include <vector> #include <map> #include <algorithm> #include <utility> #include <iomanip> #include <limits> #include <cmath> using namespace std; struct Node { vector<Node*> nb; // Node list of neighbor atoms ([Node]) int idx; // index of the atom in the residue (int) int d; // index of traverse history (int) vector<int> t; // neighbors of traverse history ([int]) string atom; // atom type (string) int res_id; // residue id of the PDB (int) vector<double> crd; // coordinate of the atom ([float,float,float]) Node(int idx_, string atom_, int res_id_, vector<double>& crd_) : idx(idx_), d(-1), atom(atom_), res_id(res_id_), crd(crd_) { } }; struct PdbData { string atom; int res_id; vector<double> crd; PdbData(string line) { stringstream ss; double d; atom = line.substr(76, 2); atom.erase(0, atom.find_first_not_of(' ')); atom.erase(atom.find_last_not_of(' ') + 1); ss << line.substr(20, 6); ss >> res_id; ss.str(""); ss.clear(); ss << line.substr(30, 8); ss >> d; crd.push_back(d); ss.str(""); ss.clear(); ss << line.substr(38, 8); ss >> d; crd.push_back(d); ss.str(""); ss.clear(); ss << line.substr(46, 8); ss >> d; crd.push_back(d); ss.str(""); ss.clear(); } }; // comparison function for sorting of traverse history class LessNeighborD { public: bool operator()(const Node* x, const Node* y) { int x_min_d, y_min_d; x_min_d = y_min_d = numeric_limits<int>::max(); for (vector<Node*>::const_iterator p = x->nb.begin(); p != x->nb.end(); ++p) { if ((*p)->d == -1) continue; x_min_d = min(x_min_d, (*p)->d); } for (vector<Node*>::const_iterator p = y->nb.begin(); p != y->nb.end(); ++p) { if ((*p)->d == -1) continue; y_min_d = min(y_min_d, (*p)->d); } return x_min_d < y_min_d; } }; class GreaterNbSize { public: bool operator()(const Node* x, const Node* y) { return x->nb.size() > y->nb.size(); } }; class LessD { public: bool operator()(const Node* x, const Node* y) { return x->d < y->d; } }; class Matching { private: // visit time of traverse history int vtime; // mapping between target and the replacing residue ((Node,Node)) vector<pair<Node*, Node*> > mapping; map<pair<string, string>, double> bond_dist; public: Matching(); double get_bond_dist(const Node* v1, const Node* v2); double distance(const Node* v1, const Node* v2); void connect(vector<Node*>& nodes); void traverse_history(Node* node); void clear_traverse_history(vector<Node*>& nodes); vector<pair<string, vector<string> > > parse_pdb(string inpdb); vector<vector<Node*> > create_nodes_list(vector<pair<string, vector<string> > >& all_lines, vector<string>& rep_res); bool match(Node* g, vector<Node*>& h, int depth); }; Matching::Matching() : vtime(0) { // table of max bond distances bond_dist[make_pair("C", "C")] = 2.0; bond_dist[make_pair("C", "N")] = 1.8; bond_dist[make_pair("C", "O")] = 1.8; bond_dist[make_pair("C", "H")] = 1.2; bond_dist[make_pair("N", "N")] = 1.8; bond_dist[make_pair("N", "O")] = 1.8; bond_dist[make_pair("H", "N")] = 1.2; bond_dist[make_pair("O", "O")] = 1.8; bond_dist[make_pair("H", "O")] = 1.2; } double Matching::get_bond_dist(const Node* v1, const Node* v2) { pair<string, string> atom_pair(make_pair(min(v1->atom, v2->atom), max(v1->atom, v2->atom))); if (bond_dist.find(atom_pair) != bond_dist.end()) return bond_dist[atom_pair]; return 0.0; } double Matching::distance(const Node* v1, const Node* v2) { double d = 0.0; for (int i = 0; i < 3; i++) { double dv = v1->crd[i] - v2->crd[i]; d += dv * dv; } return sqrt(d); } // create connect list for neighbor atoms void Matching::connect(vector<Node*>& nodes) { for (vector<Node*>::iterator p = nodes.begin(); p != nodes.end(); ++p) { for (vector<Node*>::iterator q = nodes.begin(); q != nodes.end(); ++q) { if (*p == *q || (*p)->idx > (*q)->idx) continue; if (distance(*p, *q) < get_bond_dist(*p, *q)) { (*p)->nb.push_back(*q); (*q)->nb.push_back(*p); } } } } // traverse history, similar to canonical labeling void Matching::traverse_history(Node* node) { node->d = vtime; vtime++; vector<Node*> nb; for (vector<Node*>::iterator p = node->nb.begin(); p != node->nb.end(); ++p) { if ((*p)->d == -1) nb.push_back(*p); else node->t.push_back((*p)->d); } sort(node->t.begin(), node->t.end()); sort(nb.begin(), nb.end(), LessNeighborD()); for (vector<Node*>::iterator p = nb.begin(); p != nb.end(); ++p) if ((*p)->d == -1) traverse_history(*p); } void Matching::clear_traverse_history(vector<Node*>& nodes) { for (vector<Node*>::iterator p = nodes.begin(); p != nodes.end(); ++p) { (*p)->d = -1; (*p)->t.clear(); } mapping.clear(); } vector<pair<string, vector<string> > > Matching::parse_pdb(string inpdb) { int res_id, prev_res_id = -1; vector<string> lines; vector<pair<string, vector<string> > > all_lines; string res, line; stringstream ss; fstream fs(inpdb.c_str(), ios_base::in); while (getline(fs, line)) { ss.str(""); ss.clear(); if (line.length() > 20 && line.substr(0, 4) == "ATOM") { ss << line.substr(20, 6); ss >> res_id; } else res_id = -1; if (prev_res_id == res_id) lines.push_back(line); else { if (!lines.empty()) all_lines.push_back(make_pair(res, lines)); lines.clear(); lines.push_back(line); } if (line.length() > 20 && line.substr(0, 4) == "ATOM") { res = line.substr(17, 3); res.erase(0, res.find_first_not_of(' ')); res.erase(res.find_last_not_of(' ') + 1); } else res = ""; prev_res_id = res_id; } all_lines.push_back(make_pair(res, lines)); return all_lines; } vector<vector<Node*> > Matching::create_nodes_list(vector<pair<string, vector<string> > >& all_lines, vector<string>& rep_res) { vector<vector<Node*> > nodes_list; vector<vector<PdbData> > pdb_data; for (vector<pair<string, vector<string> > >::iterator p = all_lines.begin(); p != all_lines.end(); ++p) { string res(p->first); vector<PdbData> pdbs; for (vector<string>::iterator q = p->second.begin(); q != p->second.end(); ++q) { // for l in lines if (q->length() > 78 && q->substr(0, 4) == "ATOM" && find(rep_res.begin(), rep_res.end(), res) != rep_res.end()) pdbs.push_back(PdbData(*q)); } pdb_data.push_back(pdbs); } for (vector<vector<PdbData> >::iterator p = pdb_data.begin(); p != pdb_data.end(); ++p) { if (p->empty()) continue; vector<Node*> nodes; int idx = 0; for (vector<PdbData>::iterator q = p->begin(); q != p->end(); ++q) nodes.push_back(new Node(idx++, q->atom, q->res_id, q->crd)); connect(nodes); vtime = 0; traverse_history(nodes[0]); sort(nodes.begin(), nodes.end(), LessD()); nodes_list.push_back(nodes); } return nodes_list; } bool Matching::match(Node* g, vector<Node*>& h, int depth) { for (vector<Node*>::iterator p = g->nb.begin(); p != g->nb.end(); ++p) if ((*p)->d != -1) g->t.push_back((*p)->d); if (h[depth]->t.size() != g->t.size() || h[depth]->atom != g->atom) { g->t.clear(); return false; } g->d = depth; sort(g->t.begin(), g->t.end()); if (g->t == h[depth]->t) { mapping.push_back(make_pair(h[depth], g)); if (mapping.size() == h.size()) return true; vector<Node*> nb(mapping[h[depth+1]->t[0]].second->nb); sort(nb.begin(), nb.end()); vector<Node*>::iterator end = nb.end(); for (int i = 1; i < h[depth+1]->t.size(); i++) { vector<Node*> nb2(mapping[h[depth+1]->t[i]].second->nb); sort(nb2.begin(), nb2.end()); end = set_intersection(nb.begin(), end, nb2.begin(), nb2.end(), nb.begin()); } nb.erase(end, nb.end()); sort(nb.begin(), nb.end(), GreaterNbSize()); for (vector<Node*>::iterator p = nb.begin(); p != nb.end(); ++p) if ((*p)->d == -1 && match(*p, h, depth + 1)) return true; mapping.pop_back(); } g->d = -1; g->t.clear(); return false; } int main(int argc, char* argv[]) { string inpdb, outpdb, target; vector<pair<string, string> > res_pdb_list; if (argc < 6 || (argc & 1)) { cerr << "Usage: " << argv[0] << " input_pdb output_pdb target_res replace_res1 replace_res1_pdb [replace_res2 replace_res2_pdb...]" << endl; cerr << " ex.) " << argv[0] << " input.pdb output.pdb HIS HID HID.pdb HIE HIE.pdb HIP HIP.pdb" << endl; exit(1); } inpdb = argv[1]; outpdb = argv[2]; target = argv[3]; for (int i = 4; i < argc; i += 2) res_pdb_list.push_back(make_pair(argv[i], argv[i+1])); Matching m; // calculate nodes of replacing residues map<string, vector<Node*> > rep_res; for (vector<pair<string, string> >::iterator p = res_pdb_list.begin(); p != res_pdb_list.end(); ++p) { vector<pair<string, vector<string> > > all_lines(m.parse_pdb(p->second)); vector<string> res; res.push_back(p->first); vector<vector<Node*> > nodes_list(m.create_nodes_list(all_lines, res)); rep_res[p->first] = nodes_list[0]; } // calculate nodes of target residues in the PDB vector<pair<string, vector<string> > > all_lines(m.parse_pdb(inpdb)); vector<string> res; res.push_back(target); vector<vector<Node*> > nodes_list(m.create_nodes_list(all_lines, res)); cout << "Number of " << target << ": " << nodes_list.size() << endl; // matching residues vector<string> result; bool is_found = false; for (vector<vector<Node*> >::iterator p = nodes_list.begin(); p != nodes_list.end(); ++p) { is_found = false; for (map<string, vector<Node*> >::iterator q = rep_res.begin(); q != rep_res.end(); ++q) { if (p->size() != q->second.size()) continue; m.clear_traverse_history(*p); for (vector<Node*>::iterator r = p->begin(); r != p->end(); ++r) { if (m.match(*r, q->second, 0)) { cout << target << " " << setw(4) << (*r)->res_id << " -> " << q->first << endl; result.push_back(q->first); is_found = true; break; } } if (is_found) break; } if (!is_found) { cout << target << " " << setw(4) << (*p)[0]->res_id << " -> Not found." << endl; result.push_back(target); } } // write PDB fstream fs(outpdb.c_str(), ios_base::out); int idx = 0; for (vector<pair<string, vector<string> > >::iterator p = all_lines.begin(); p != all_lines.end(); ++p) { if (p->first == target) { for (vector<string>::iterator q = p->second.begin(); q != p->second.end(); ++q) { size_t pos = q->find(target); if (pos != string::npos) q->replace(pos, target.size(), result[idx]); fs << *q << endl; } idx++; } else { for (vector<string>::iterator q = p->second.begin(); q != p->second.end(); ++q) fs << *q << endl; } } return 0; }
コメント